Sometimes surgeons ask to have nonmedical devices sterilized, such as spoons, hockey pucks, and fish hooks. Healthcare staff may want to accommodate their requests, but there are times when they cannot do so. A central service (CS) should only sterilize medical devices that have undergone validation testing, which demonstrates that they are safe to undergo sterilization and thus are safe for patient use.
The terms "validation" and "verification" testing are sometimes used interchangeably when referring to processing of medical devices, but they are very different processes. Healthcare facilities perform verification testing, whereas device manufacturers perform validation testing. Both processes are important to ensure proper sterility, and sterile processing professionals should be familiar with the steps involved in each.
Validation: A scientific process
The device manufacturer performs validation–a documented procedure consisting of scientifically valid methods to obtain, record, and interpret the results. The purpose is to establish that a process will consistently yield a medical device complying with predetermined specifications.
Before a reusable medical device is introduced to the medical community, the manufacturer must obtain clearance from the US Food and Drug Administration (FDA). To do so, the manufacturer must demonstrate that the medical device can be safely and effectively reprocessed between patient uses.
The reuse of a medical device is associated with the risk of disease transmission from one patient to another or from environmental sources to a patient, and with the risk of inadequate device performance after reprocessing. Manufacturers of reusable medical devices must provide instructions for use (IFU) for reusable medical devices that explain how to handle, clean, disinfect, test, package, sterilize and, if applicable, aerate their products. Manufacturers also have to conduct and document any testing necessary to validate the suitability of these instructions.
Guidelines
These obligations are outlined in FDA labeling regulations (21 CFR 801), and recommendations are published in the FDA guidance document, Reprocessing Medical Devices in Health Care Settings: Validation Methods and Labeling Guidance for Industry and Food and Drug Administration Staff, issued on March 17, 2015; Appendix E, updated on June 9, 2017.
The FDA defines reprocessing as validated processes that render a used or contaminated medical device ready for the next use. The processes must be designed to remove soil and contaminants by cleaning and to inactivate microorganisms by disinfection or sterilization.
Validation studies are performed in accordance with AAMI TIR 12, Designing, Testing, and Labeling Reusable Medical Devices for Reprocessing in Health Care Facilities: A Guide for Medical Device Manufacturers, and AAMI TIR 30, A Compendium of Processes, Materials, Test Methods, and Acceptance Criteria for Cleaning Reusable Medical Devices. These documents describe designs that can help medical device manufacturers ensure that their products are safely and effectively reprocessed, as well as a summary of test methods available in the published literature.
Information about common decontamination, cleaning, disinfection, and sterilization processes helps manufacturers develop and validate reprocessing procedures that are used in healthcare facilities. Information about the test methods helps them develop reprocessing IFU and demonstrate that each device can be thoroughly cleaned without damage or remaining residues.
Cleaning protocols

Cleaning validation testing in a validation lab, inoculating an instrument with a soil simulation to represent the challenges in proper cleaning and reprocessing of devices. The lock box is the most difficult to clean. Photo courtesy Nelson Labs, Salt Lake City. Used with permission.
Cleaning validation activities are based on all-inclusive validation protocols that use soil simulations to represent the challenges in proper cleaning and reprocessing of devices. These include:
- implementation of the cleaning process
- medical devices that represent the worst case, ie, devices that are the most contaminated and most challenging to reprocess
- at least two quantitative test methods that are as close as possible to the soil in actual use.
Cleaning validation protocols specify predetermined cleaning test endpoints. These protocols are designed to establish that the most inaccessible locations on the devices can be adequately cleaned during routine processing. The simulated soil represents the worst-case scenarios of exposure to contaminants that will result from the least thorough cleaning method.
A specific type of artificial soil is used to inoculate (inject) into the device all locations likely to contact patient materials, including all locations that are hard to clean. If a device has features that can accumulate soil with repeated use, the validation studies will use devices that have undergone some simulated use. These studies incorporate multiple full-use cycles because medical devices should be designed to clean all soil that accumulates over time. All operative techniques of the device, such as repeated articulations, flexures, and/or manipulations, are also inoculated and tested under worst-case conditions.
If the use of a device is likely to repeatedly "push" soil into a hard-to-reach area, the validation testing should include repeated soiling to reproduce a worst-case situation. Likewise, a device may not be cleaned immediately after use, and soil may dry on it. Validation methods will allow soils to dry for a certain period to simulate the worst case or longest duration of time. Cleaning validation protocols should use the shortest times, lowest temperatures, weakest dilutions, etc, for each step of the cleaning process.
Devices are then subjected to a validated method of extraction for recovery of residual soil.
The extraction method removes the testing soil that was placed on all external and internal surfaces and mated surfaces, where it is most difficult to remove. Some devices have complex internal structures (eg, lumens or internal moving parts) that may become soiled during use and are hard to access during cleaning and extraction. Therefore, cleaning methods–including disassembly–are designed to access these surfaces for cleaning and sterilization.
Medical device manufacturers do not validate cleaning solutions. Cleaning solution manufacturers are responsible for providing information about the water quality, cleaning agent concentration, exposure time, and other parameters necessary for effective use of their products.
Sterilization validation testing
Medical device manufacturers conduct validation testing in sterilizers that are available to healthcare facilities and that use steam, ethylene oxide, hydrogen peroxide, dry heat, and ozone. Medical devices are packaged in an FDA-cleared packaging system that provides a microbial barrier to maintain sterility prior to use.
To qualify sterilization cycle parameters, the device manufacturer must show that the cycle has sufficient lethality to produce the desired sterility assurance level for the device. An "overkill" approach is used to demonstrate cycle lethality because the pre-sterilization bioburden of reusable devices processed in healthcare facilities is not routinely recognized.
The overkill sterilization method is based on the concept that it will be able to inactivate a resistant microbiological challenge and provide an additional safety factor. An example of cycle overkill is a 6-log reduction in half of the steam sterilization cycle exposure time of a microbiological challenge.
Steam sterilization, the most widely used sterilant in healthcare facilities, demonstrates how sterilization validation works. The test uses preparations of Geobacillus stearothermophilus containing a population of 106 spores, which are the most resistant to steam sterilization. The medical device is inoculated in the most resistant yet accessible locations. The medical device being validated for steam sterilization is subjected to a half-cycle. If the validated sterilization cycle is 4 minutes at 270 ºF, the instrument is inoculated with Geobacillus stearothermophilus in areas most resistant to the sterilization cycle and then sterilized for 2 minutes. At the end of the steam sterilization cycle, the instrument is cultured in the most challenging areas for the sterilant contact. The culture should show no growth, thus demonstrating that the medical device is sterile.
After the sterilization process is completed, the manufacturer demonstrates the physical and functional compatibility of devices with the sterilization process, taking into account the intended use of the device and the effects of repeated sterilization.
Material properties–such as physical strength and dimensions, resilience, and permeability (for gas sterilization)–are evaluated after multiple sterilization cycles. The device is checked for material effects such as crazing, cracking, embrittlement, and phase separation, which can degrade polymeric materials. Discoloration, staining, and other negative aesthetic effects are also identified. All movable or hinged parts are opened and closed when inspecting the device for visible soil and ease of movement.
A sterilization residue check detects substances that can be harmful to patients and sometimes to staff. Residues are often difficult to see, so the test is performed by a residue reagent test and/or tactile feedback that can reveal sticky or oily surfaces and stiff hinges. Devices are checked to ensure that markings such as depth or size can be easily seen and are not dull or rubbed off. There is also a test to determine if processing in accordance with the provided instructions will harm the device and thus limit its useful life.
Verification testing
Healthcare facilities perform verification tests to ensure that they can duplicate the manufacturer's recommended cleaning and sterilization methods and that the instructions are followed. Verification entails performing documented procedures to obtain, record, and interpret the results required to establish that sterilization specifications have been met.
When a medical device is introduced into a healthcare facility, cleaning verification should be performed to ensure that the device can be thoroughly cleaned. Developing a cleaning verification involves establishing, clarifying, and documenting a standard cleaning process based on published and validated recommended practices or guidelines and the IFU.
The cleaning verification process includes critical steps that are verified through comprehensive training and observation. All visible organic soil and contamination must be removed, and CS technicians must demonstrate the correct cleaning process without variation. Process controls should be in place to ensure complete and consistent cleaning.
Policies and procedures should be consistent with standards and recommended practices, including those of the device and detergent manufacturers and, if used, those of the manufacturer of the automatic processing equipment.
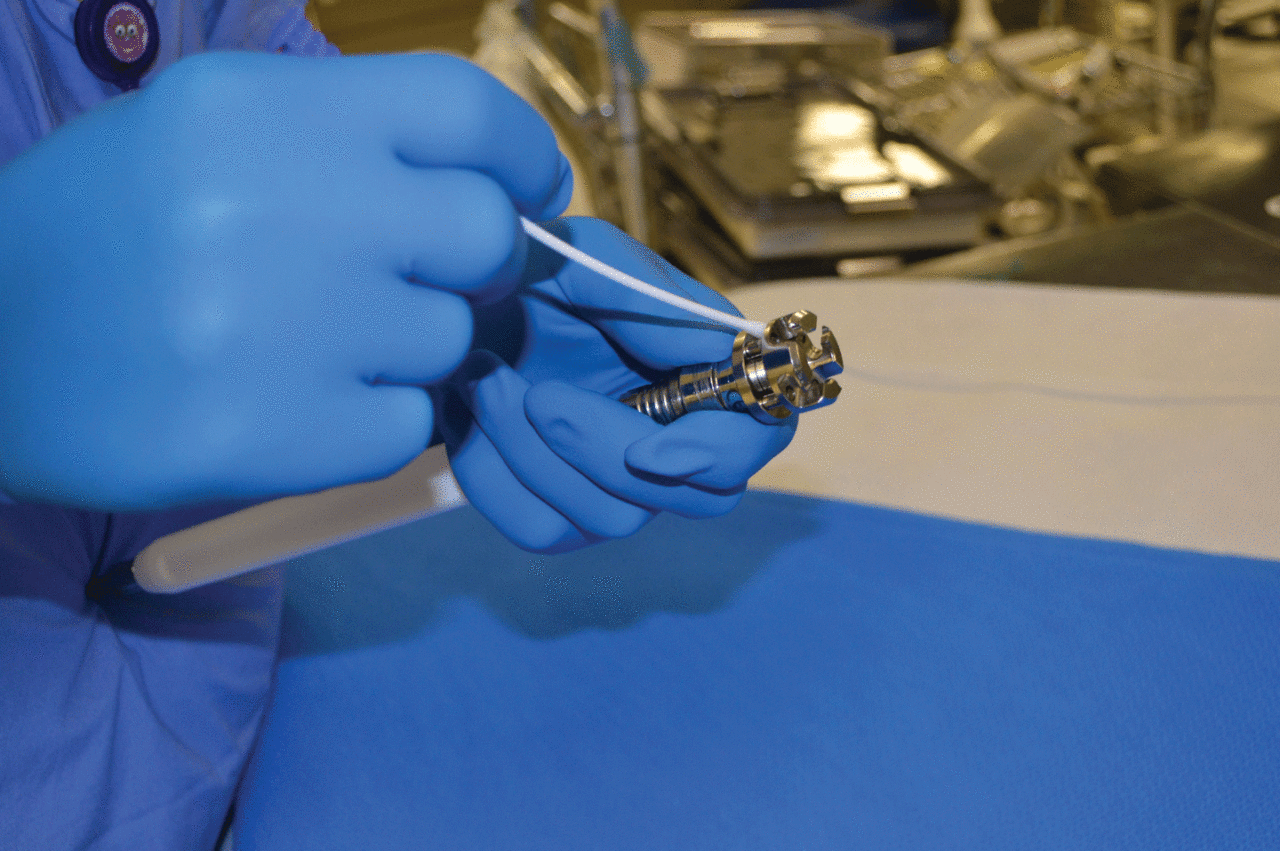
Cleaning verification using an adenosine triphosphate (ATP) test.
To confirm efficacy of the device cleaning process, an inspection is performed–either visually or with the use of enhanced visualization tools such as lighted magnification or borescopes to identify residues not seen by the unaided eye. Commercial test kits are also available to measure organic residues such as adenosine triphosphate (ATP), protein, and hemoglobin, which cannot be detected using visual inspection. Cleaning verification tests should be used according to their IFU.
A functionality check should be done to ensure the device operates as it was intended to. This may be defined by the device manufacturer and is a part of the basic inspection, preparation, and packaging. The device manufacturer may provide tests to check for debris and residue in areas that cannot be visually inspected.
Product testing, which verifies that the device can be sterilized in a healthcare facility, is not a substitute for validation. To perform product testing in healthcare settings, chemical indicators (CIs) and biological indicators (BIs) validated for that method of sterilization are used to verify that the sterilant provided the required lethality to yield a sterile product. These indicators are placed strategically throughout the test pack at the points most difficult to sterilize (those that are most resistant to steam penetration).
Not all medical devices need to be tested. Instead, product "families" of devices or instrument sets that represent the most challenges in sterilization are tested. The concept of product families enables the healthcare facility to ensure a high level of sterility assurance without testing all products being sterilized.
For verification testing, each test pack should have BIs and CIs placed in the most difficult area to sterilize. The number of indicators, based on the size and configuration of the test pack, should be placed in areas representing the greatest sterilization challenge, and the location should be documented on the indicators. When the sterilization cycle is complete, the locations and test results should be recorded. A digital camera can be used for additional documentation.
Product test samples are packaged as normal, labeled "test pack," and processed in a routine load with other items. Test packs are placed throughout the sterilizer load in the areas that are most difficult to sterilize or in locations that would be most resistant to steam sterilization, such as over the drain.
Before and during product testing, all sterilizer quality monitors are checked, including physical monitors, CIs, and BIs. As with all sterilization loads, the physical parameters are reviewed to ensure the correct cycle was selected and that they were met.
As soon as the sterilization process is complete, the test pack is opened, the contents are checked for moisture, CIs are reviewed to meet their endpoint, and BIs are incubated. The test packs are inspected for moisture, and the contents of the test sample should be either reprocessed or discarded.
Unacceptable test results (positive BIs, CIs that have not reached their endpoint, and/or evidence of moisture) must be investigated. Documentation includes the test protocol, placement of BIs and CIs, the test results, and any corrective actions that were taken.
Remember who does what and why
Healthcare facilities perform verification studies of medical devices, using available quality monitors. Manufacturers perform validation testing, which involves specific scientific measurement analysis with microbiological, engineering, toxicological, and sometimes clinical evaluations to demonstrate the medical device can be effectively cleaned and sterilized.
All of a manufacturer's IFU involved in processing medical devices must be followed. Healthcare facilities verify that the medical devices validated by manufacturers can be cleaned and sterilized in their facilities. Because healthcare facilities cannot perform the required validation studies, they cannot validate items brought in for sterilization.

Susan Klacik, BS, CRCST, CHL, CIS, ACE, FCS, is a clinical educator for IAHCSMM (International Association of Healthcare Central Service Materiel Management) and a representative to AAMI. She also serves on AORN's Guidelines Advisory Board.
References
Association for the Advancement of Medical Instrumentation. A compendium of processes, materials, test methods, and acceptance criteria for cleaning reusable medical devices. AAMI TIR 30:2016. www.aami.org.
Association for the Advancement of Medical Instrumentation. Comprehensive guide to steam sterilization and sterility assurance in health care facilities. ANSI/AAMI ST79:2017. www.aami.org.
Association for the Advancement of Medical Instrumentation. Designing, testing, and labeling reusable medical devices for reprocessing in health care facilities. AAMI TIR 12:2010. www.aami.org.
Food and Drug Administration. Reprocessing medical devices in health care settings: Validation methods and labeling guidance for industry and Food and Drug Administration staff. Issued on March 17, 2015. Appendix E, updated on June 9, 2017. https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm253010.pdf.